Intrinsic Acute Kidney Injury: Structural Nephron Injury
Intrinsic acute kidney injury refers to acute kidney injury caused by direct structural damage to the nephron, most commonly affecting the renal tubules, interstitium, glomeruli, or renal microvasculature. Unlike pre-renal AKI, intrinsic AKI reflects true tissue injury rather than functional adaptation to reduced perfusion. Intrinsic AKI can explain why kidney dysfunction persists despite restoration of blood flow, why urine findings change, and why recovery depends on cellular repair rather than haemodynamic correction alone.
What You Need to Know
Intrinsic acute kidney injury occurs when the structures of the nephron itself become damaged. Unlike pre-renal AKI, where reduced blood flow temporarily limits filtration, intrinsic AKI reflects direct injury to the filtering and tubular machinery of the kidney. Once nephron cells are damaged, the kidney loses not only filtration capacity but also its ability to regulate electrolytes, acid–base balance, and fluid composition.
The most common form of intrinsic AKI is acute tubular necrosis (ATN), in which tubular epithelial cells are injured by prolonged ischaemia, toxins, or severe inflammation. These cells have very high energy demands, so even brief reductions in oxygen delivery or exposure to nephrotoxic substances can disrupt cellular metabolism, leading to cell swelling, detachment, and death. As injured cells slough into the tubular lumen, they obstruct flow and allow filtrate to leak back into the interstitium, meaning that glomerular filtration may occur but effective urine formation falls.
Because the injury is structural rather than purely functional, intrinsic AKI does not rapidly improve when blood flow is restored. Creatinine continues to rise, urine composition becomes abnormal, and electrolyte and acid–base disturbances worsen. This is why intrinsic AKI is associated with hyperkalaemia, metabolic acidosis, and uraemia even when urine output appears adequate.
Intrinsic AKI also triggers an inflammatory response within the kidney. Cytokines, endothelial injury, and microvascular dysfunction impair oxygen delivery to surviving nephrons, creating a self-perpetuating cycle of injury. This means the kidney becomes both a target and a driver of systemic illness, contributing to multi-organ dysfunction in critically ill patients.
Clinically, intrinsic AKI is suspected when kidney function fails to improve after optimisation of perfusion or relief of obstruction. It is often preceded by a pre-renal state that was not corrected in time, or it may occur after direct nephrotoxic exposure or severe sepsis.
Common causes include:
prolonged hypotension or shock leading to ischaemic injury
nephrotoxic drugs (e.g. aminoglycosides, contrast, chemotherapy)
myoglobin or haemoglobin from rhabdomyolysis or haemolysis
severe sepsis and systemic inflammation
Intrinsic AKI therefore represents a transition from reversible functional stress to true kidney damage, and recovery depends on the ability of surviving tubular cells to regenerate and restore nephron integrity.
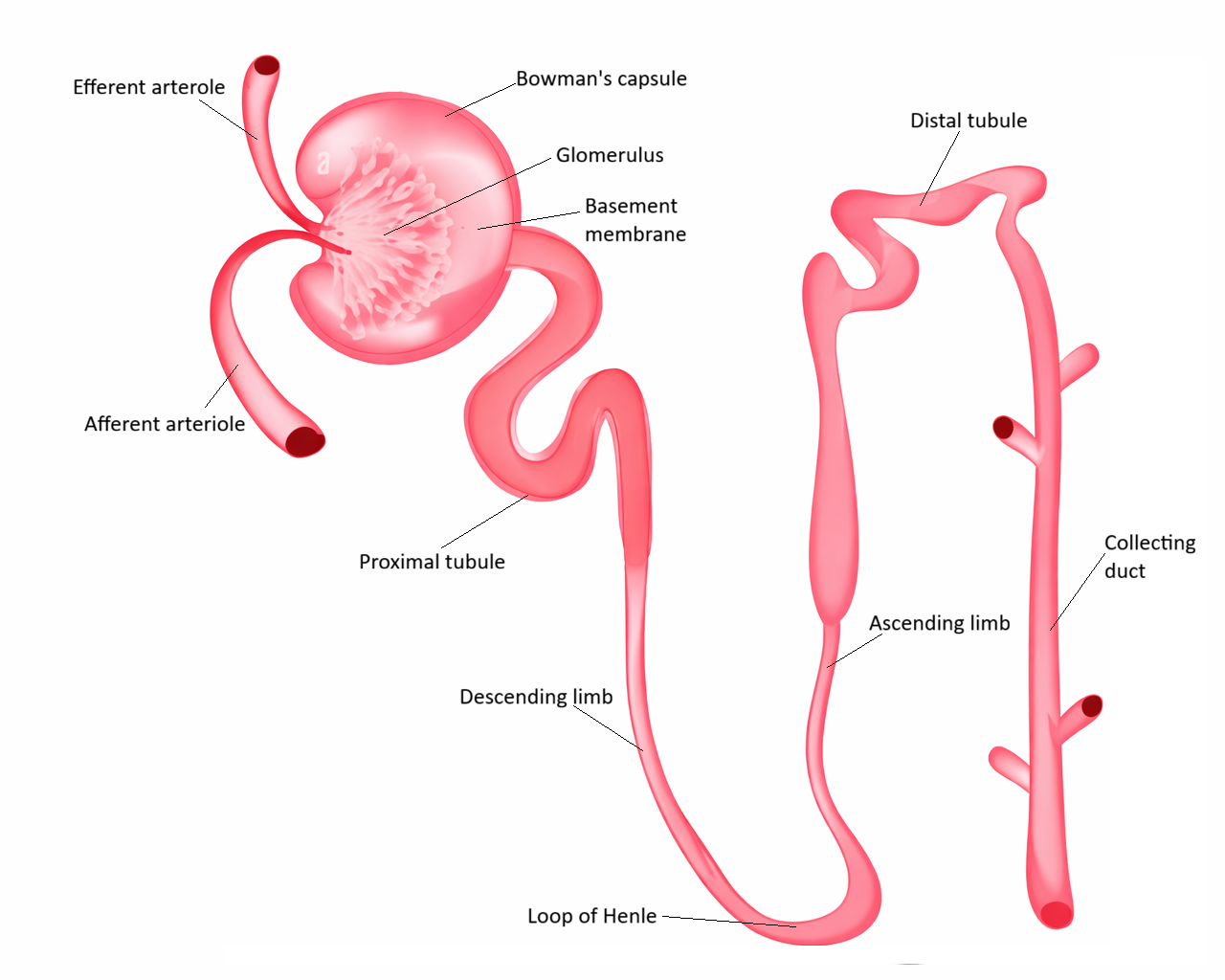
Image: The nephron is the functional unit of the kidney, responsible for filtration, reabsorption, and secretion. In intrinsic kidney injury, damage occurs within these structures, disrupting normal function and impairing the kidney’s ability to maintain fluid, electrolyte, and waste balance.
Beyond the Basics
Tubular Cell Injury and Loss of Polarity
Renal tubular epithelial cells are among the most metabolically active cells in the body because they must continuously transport sodium, water, and solutes against concentration gradients. This makes them highly dependent on a steady oxygen and ATP supply. During ischaemia or exposure to nephrotoxins, ATP depletion disrupts ion pumps, cytoskeletal integrity, and intracellular signalling, causing the cells to swell, lose polarity, and detach from their anchoring basement membrane.
As these injured cells slough into the tubular lumen, they form obstructive casts that block the flow of filtrate. This obstruction raises intratubular pressure and pushes back against glomerular filtration, reducing net filtration despite preserved blood flow. At the same time, loss of epithelial polarity prevents normal reabsorption of sodium and water, producing urine that is relatively dilute and sodium-rich, a hallmark of intrinsic tubular injury.
Back-Leak of Filtrate and Ineffective Urine Formation
In a healthy nephron, tubular epithelial cells form a tightly regulated barrier that ensures filtrate moves in one direction—from the glomerulus to the collecting system and out of the body. In intrinsic AKI, disruption of tight junctions and epithelial integrity allows filtrate to escape from the tubules into the interstitium and peritubular capillaries, effectively re-entering the circulation.
This back-leak creates a situation where filtration may still occur at the glomerulus, but very little of that filtrate is converted into effective urine. Creatinine and waste products therefore accumulate even when renal blood flow appears adequate. Clinically, this explains why oliguria and rising urea and creatinine may persist after haemodynamic correction and why intrinsic AKI cannot be reversed simply by restoring perfusion.
Inflammatory Amplification and Microvascular Dysfunction
Intrinsic AKI is now recognised as an active inflammatory disease rather than a passive consequence of injury. Damaged tubular cells release cytokines, chemokines, and danger-associated molecular signals that recruit neutrophils and macrophages into the renal interstitium. Endothelial cells become activated, increasing vascular permeability and promoting leukocyte adhesion.
These changes disrupt the delicate microcirculation of the kidney. Capillary swelling, congestion, and microthrombi reduce oxygen delivery to surviving nephrons, creating patchy but severe tissue hypoxia even when systemic blood pressure is normal. This microvascular failure allows kidney injury to spread beyond the original insult and explains why AKI can worsen despite correction of the triggering event.
Nephrotoxicity and Direct Cellular Injury
Many cases of intrinsic AKI arise not from reduced perfusion but from direct chemical or biological toxicity. Drugs such as aminoglycosides, chemotherapy agents, NSAIDs, and iodinated contrast injure tubular cells through oxidative stress, mitochondrial damage, and disruption of intracellular calcium regulation. Endogenous toxins released during muscle breakdown, haemolysis, or tumour lysis exert similar effects.
Because these injuries act directly on renal cells, kidney failure can develop even when blood pressure and volume status appear stable. This explains why critically ill patients may develop AKI after imaging, medication exposure, or massive tissue injury and highlights why nephrotoxin avoidance is central to renal protection in vulnerable patients.
Repair, Regeneration, and Incomplete Recovery
Unlike most epithelial tissues, renal tubules retain a limited capacity for regeneration after injury. Surviving tubular cells can dedifferentiate, migrate, proliferate, and re-establish a functional epithelial lining, allowing partial or even full recovery of kidney function over days to weeks. During this period, however, filtration and regulatory capacity remain impaired, leaving patients vulnerable to fluid, electrolyte, and metabolic instability.
When injury is severe, prolonged, or recurrent, the repair process becomes maladaptive. Instead of normal tubular regeneration, fibroblasts are activated and extracellular matrix accumulates, replacing functional nephron tissue with scar. This permanent loss of nephron mass explains why intrinsic AKI is one of the strongest risk factors for future chronic kidney disease and why each episode leaves the kidneys more vulnerable than before.
Clinical Connections
Intrinsic AKI typically presents with persistent oliguria or non-oliguric renal failure accompanied by rising creatinine, urea, and abnormal urine chemistry. Because the injury lies within the nephron itself, kidney function does not improve rapidly with fluid resuscitation, distinguishing it from pre-renal states where perfusion alone restores filtration. Urine is often dilute and sodium-rich, reflecting tubular damage and loss of reabsorptive capacity, and microscopic analysis may reveal granular or epithelial cell casts that directly reflect tubular breakdown.
Clinically, intrinsic AKI is most often seen in the context of severe sepsis, prolonged hypotension, major surgery, trauma, rhabdomyolysis, contrast exposure, or drug toxicity. Patients may initially appear haemodynamically stable, yet kidney injury continues to progress because inflammation, microvascular dysfunction, and epithelial injury persist inside the kidney even after systemic circulation is corrected. This is why creatinine may continue to rise for several days after the original insult has resolved.
Management is therefore protective and supportive rather than corrective. The goal is to preserve remaining nephron function while allowing time for cellular repair and regeneration to occur. Over-resuscitation with fluids can worsen renal congestion and delay recovery, while nephrotoxic medications or contrast exposure can convert partial injury into irreversible damage.
In practice this means clinicians closely monitor for:
Failure of creatinine to fall after haemodynamic correction
Persistent or worsening oliguria despite adequate perfusion
Rising potassium, acidosis, and fluid overload
Urine sodium and sediment suggesting tubular injury
Patients with intrinsic AKI often require careful balancing of fluids, electrolytes, and medications while awaiting renal recovery. Dialysis may be needed temporarily if metabolic or volume derangements become life-threatening, but recovery depends primarily on the extent of tubular survival and the absence of further insults. Early recognition and avoidance of secondary injury are therefore the most powerful tools for preserving long-term kidney function.
Concept Check
Why does intrinsic AKI persist despite restoration of renal perfusion?
How does tubular cell injury reduce effective urine formation?
What is meant by back-leak of filtrate, and why does it matter?
How does inflammation perpetuate renal injury after the initial insult?
Why does intrinsic AKI increase the risk of chronic kidney disease?