CYSTIC FIBROSIS
Cystic fibrosis (CF) is an inherited, multisystem disorder characterised by abnormal chloride and sodium transport across epithelial surfaces. In the respiratory system, this defect results in thick, dehydrated airway secretions that impair mucociliary clearance and predispose the lungs to chronic infection and inflammation. Over time, repeated cycles of infection and immune response cause progressive structural lung damage and respiratory failure.
Although cystic fibrosis affects multiple organs, respiratory complications remain the leading cause of morbidity and mortality. Understanding the underlying pathophysiology explains why airway obstruction, infection and inflammation are persistent and progressive features of the disease.
What You Need to Know
Cystic fibrosis is a genetic disorder caused by mutations in the CFTR gene, which encodes a chloride channel expressed on epithelial cell membranes in the airways, gastrointestinal tract, pancreas, and other organs. In the respiratory system, CFTR plays a critical role in regulating salt and water movement across the airway epithelium. When CFTR function is defective, chloride secretion into the airway lumen is reduced while sodium absorption is increased. Water follows sodium, resulting in dehydration of the airway surface liquid that normally keeps mucus thin and mobile.
Dehydrated airway secretions become thick, sticky, and difficult to clear. The cilia lining the airways are unable to move this viscous mucus effectively, leading to impaired mucociliary clearance. As mucus accumulates, it obstructs small airways and traps inhaled bacteria. Several key processes drive early lung disease in cystic fibrosis:
Airway surface dehydration, producing abnormally thick mucus
Impaired mucociliary clearance, allowing secretions to stagnate
Early bacterial colonisation, particularly with organisms adapted to thick mucus environments
Persistent mucus retention creates an ideal environment for chronic infection and ongoing inflammation. Neutrophils are recruited to the airways in large numbers, releasing proteases and inflammatory mediators that damage airway walls. Over time, repeated cycles of obstruction, infection, and inflammation lead to bronchiectasis, progressive airflow limitation, and declining lung function. Respiratory disease is therefore the major determinant of morbidity and mortality in cystic fibrosis, reflecting the central role of abnormal airway secretions in driving long-term lung damage.
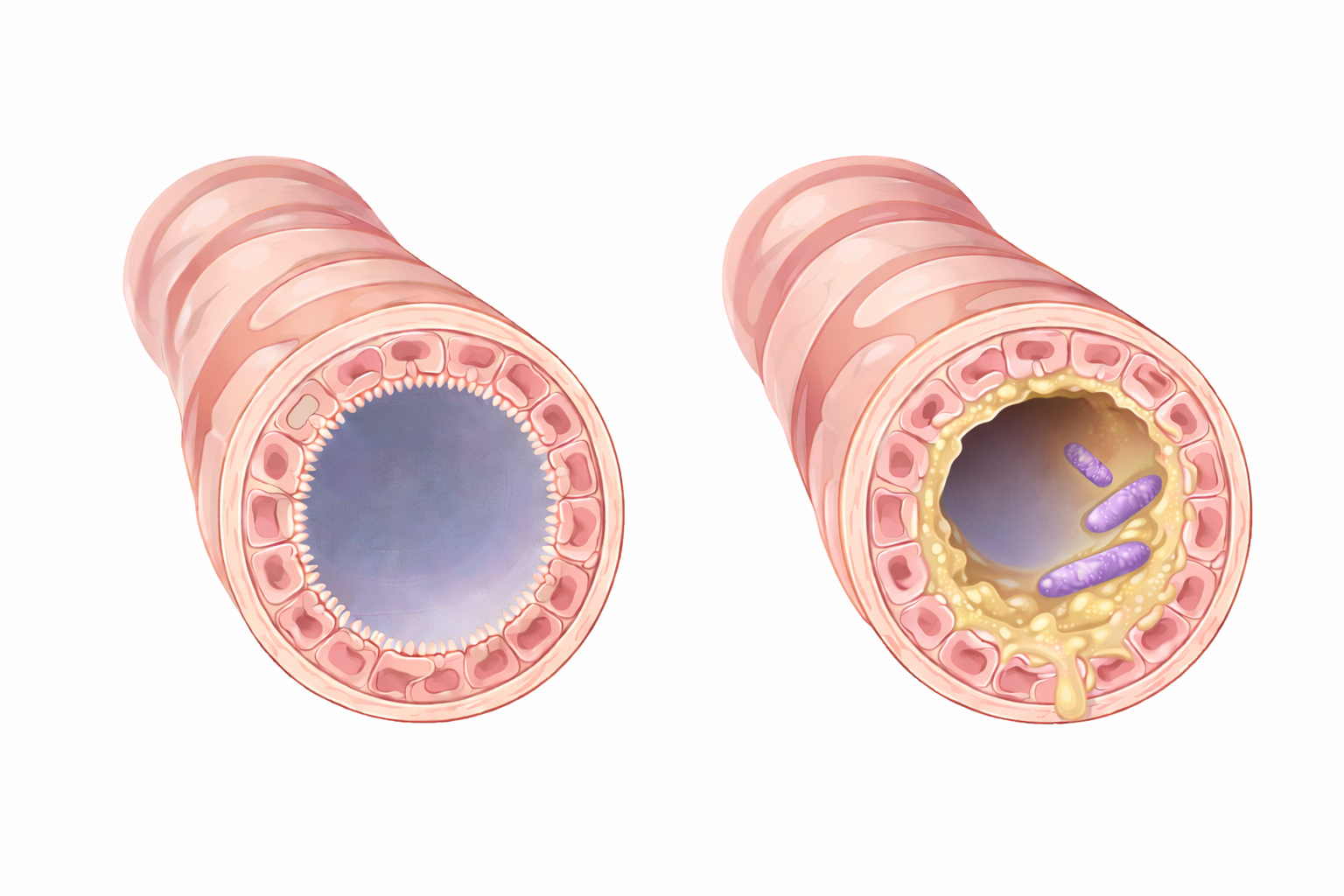
Image: Left: a normal bronchiole with a clear, open lumen allowing airflow. Right: a bronchiole obstructed by thick mucus within the airway, narrowing the lumen and impairing airflow.
Beyond the Basics
Ion Transport Dysfunction and Airway Dehydration
In healthy airways, a balance between chloride secretion and sodium absorption maintains a thin, hydrated layer of airway surface liquid that allows cilia to beat effectively. In cystic fibrosis, defective CFTR function disrupts this balance. Chloride secretion into the airway lumen is reduced, while sodium absorption is excessive. Water follows sodium back into epithelial cells, leaving the airway surface dehydrated. Mucus becomes thick and sticky, adheres to airway walls, and cannot be mobilised by ciliary action. As a result, normal clearance of inhaled particles and microorganisms is severely compromised from early life.
Mucus Obstruction and Airflow Limitation
As dehydrated mucus accumulates, it progressively narrows and obstructs the airways, particularly the smaller bronchioles. This increases airway resistance and produces an obstructive pattern of lung disease. During expiration, narrowed and weakened airways may collapse prematurely, trapping air distal to the obstruction. Air trapping leads to hyperinflation, increased work of breathing, and reduced ventilatory efficiency. With repeated episodes of obstruction and infection, airway injury becomes cumulative and increasingly difficult to reverse.
Chronic Infection and Inflammatory Injury
Impaired mucociliary clearance allows bacteria to persist within the airway mucus. Organisms such as Staphylococcus aureus and Pseudomonas aeruginosa are particularly well adapted to this environment and commonly establish chronic colonisation. The immune response is dominated by neutrophils, which migrate into the airways in large numbers. These cells release proteases, oxidants, and inflammatory mediators intended to control infection. However, this inflammatory response also damages airway epithelium, degrades connective tissue, and destroys elastic support, accelerating structural lung injury.
Development of Bronchiectasis
Repeated cycles of mucus obstruction, infection, and inflammation weaken the bronchial walls, leading to bronchiectasis. Affected airways become permanently dilated and lose their ability to clear secretions effectively. Dilated bronchi trap even larger volumes of mucus, further promoting infection and inflammation. This creates a self-perpetuating cycle in which structural damage drives ongoing disease progression. Once bronchiectasis is established, the changes are irreversible and represent a major contributor to long-term respiratory decline.
Gas Exchange Impairment and Respiratory Failure
As airway obstruction worsens and parenchymal damage accumulates, ventilation becomes increasingly uneven. Some lung regions are poorly ventilated but continue to receive blood flow, leading to ventilation–perfusion mismatch and progressive hypoxaemia. In advanced disease, respiratory muscles must work harder to overcome airflow limitation and hyperinflation, increasing the risk of fatigue. Carbon dioxide retention may develop as ventilation becomes inadequate, producing mixed Type 1 and Type 2 respiratory failure. Chronic hypoxaemia can also drive pulmonary vascular changes, leading to pulmonary hypertension and eventual right-sided heart failure as late complications of cystic fibrosis lung disease.
Clinical Connections
Cystic fibrosis commonly presents with chronic cough, recurrent chest infections, wheeze, and gradually worsening dyspnoea as thick airway secretions obstruct airflow and promote persistent infection. Sputum is often thick and difficult to clear due to dehydration of the airway surface liquid, allowing mucus to accumulate within the bronchi and bronchioles. Exacerbations occur when bacterial load and inflammation increase, leading to acute deterioration in symptoms and lung function. Over time, repeated airway obstruction and infection reduce exercise tolerance and may lead to increasing oxygen requirements, particularly during exertion or intercurrent illness.
Several clinical features arise directly from the underlying airway pathology:
Frequent infective exacerbations, due to impaired mucus clearance and chronic bacterial colonisation
Progressive airflow limitation, caused by mucus plugging, airway wall damage, and bronchiectasis
Exertional hypoxaemia, resulting from uneven ventilation and ventilation–perfusion mismatch
Pulmonary function testing typically shows an obstructive pattern, with declining FEV₁ as airway narrowing, air trapping, and loss of elastic recoil progress. Serial reductions in FEV₁ correlate closely with cumulative structural lung damage and disease severity. Imaging commonly demonstrates mucus plugging, airway wall thickening, and established bronchiectasis, all of which indicate irreversible airway change. These findings explain the progressive nature of respiratory decline in cystic fibrosis and why early recognition of deterioration and aggressive management of infection and mucus burden are essential to slowing lung function loss.
Concept Check
How does CFTR dysfunction lead to thickened airway secretions?
Why is mucociliary clearance impaired in cystic fibrosis?
How do chronic infection and inflammation contribute to bronchiectasis?
Why does cystic fibrosis produce an obstructive pattern of lung disease?
How can cystic fibrosis progress to mixed respiratory failure?