ATHEROSCLEROSIS & CORONARY ARTERY DISEASE
Atherosclerosis is a chronic, slowly progressive inflammatory disease affecting medium and large arteries. It results from endothelial injury, lipid accumulation and maladaptive immune responses that lead to plaque formation within arterial walls. When atherosclerosis affects the coronary arteries, it produces coronary artery disease (CAD), the leading cause of myocardial ischaemia, angina and myocardial infarction.
Atherosclerosis is not simply “fat clogging arteries.” It is a dynamic pathological process involving endothelial dysfunction, lipid oxidation, smooth muscle proliferation, chronic inflammation and structural remodelling. The stability of a plaque, rather than its size alone, determines the risk of acute coronary events. Understanding these mechanisms is essential for recognising the spectrum of CAD presentations and their underlying pathophysiology.
What You Need to Know
Atherosclerosis is a chronic inflammatory disease of the arterial wall and the underlying cause of most coronary artery disease. It begins with endothelial dysfunction, where the normally protective inner lining of the artery loses its ability to regulate vascular tone, inhibit inflammation, and prevent thrombosis. Damage from factors such as hypertension, smoking, diabetes, and dyslipidaemia increases endothelial permeability and reduces nitric oxide availability, impairing vasodilation and promoting inflammation. Low-density lipoprotein (LDL) particles enter the arterial intima, become oxidised, and trigger an immune response that drives plaque formation.
Monocytes adhere to the injured endothelium, migrate into the vessel wall, and differentiate into macrophages. These macrophages ingest oxidised LDL, becoming foam cells that accumulate and form fatty streaks, the earliest visible lesions of atherosclerosis. Although initially asymptomatic, these changes mark the beginning of a progressive process that can evolve over decades. As inflammation persists, the arterial wall becomes a site of ongoing immune activation rather than a passive conduit for blood flow.
Key stages in plaque development include:
Endothelial dysfunction and lipid infiltration, allowing LDL to enter and oxidise within the intima.
Foam cell and fatty streak formation, driven by macrophage uptake of oxidised LDL.
Fibrous plaque development, with smooth muscle proliferation and extracellular matrix deposition forming a cap over a lipid-rich core.
As plaques enlarge, they narrow the coronary artery lumen and limit the ability to increase blood flow during periods of increased myocardial oxygen demand, such as exercise or stress. This supply–demand mismatch produces ischaemia and symptoms of stable angina. More dangerous than gradual narrowing, however, is plaque instability. Thin or inflamed fibrous caps are prone to rupture, exposing highly thrombogenic material to circulating blood. This triggers acute thrombus formation, which may partially or completely occlude the vessel. Sudden coronary occlusion is the pathophysiological basis of acute coronary syndromes, including myocardial infarction, and represents the most life-threatening consequence of atherosclerosis.
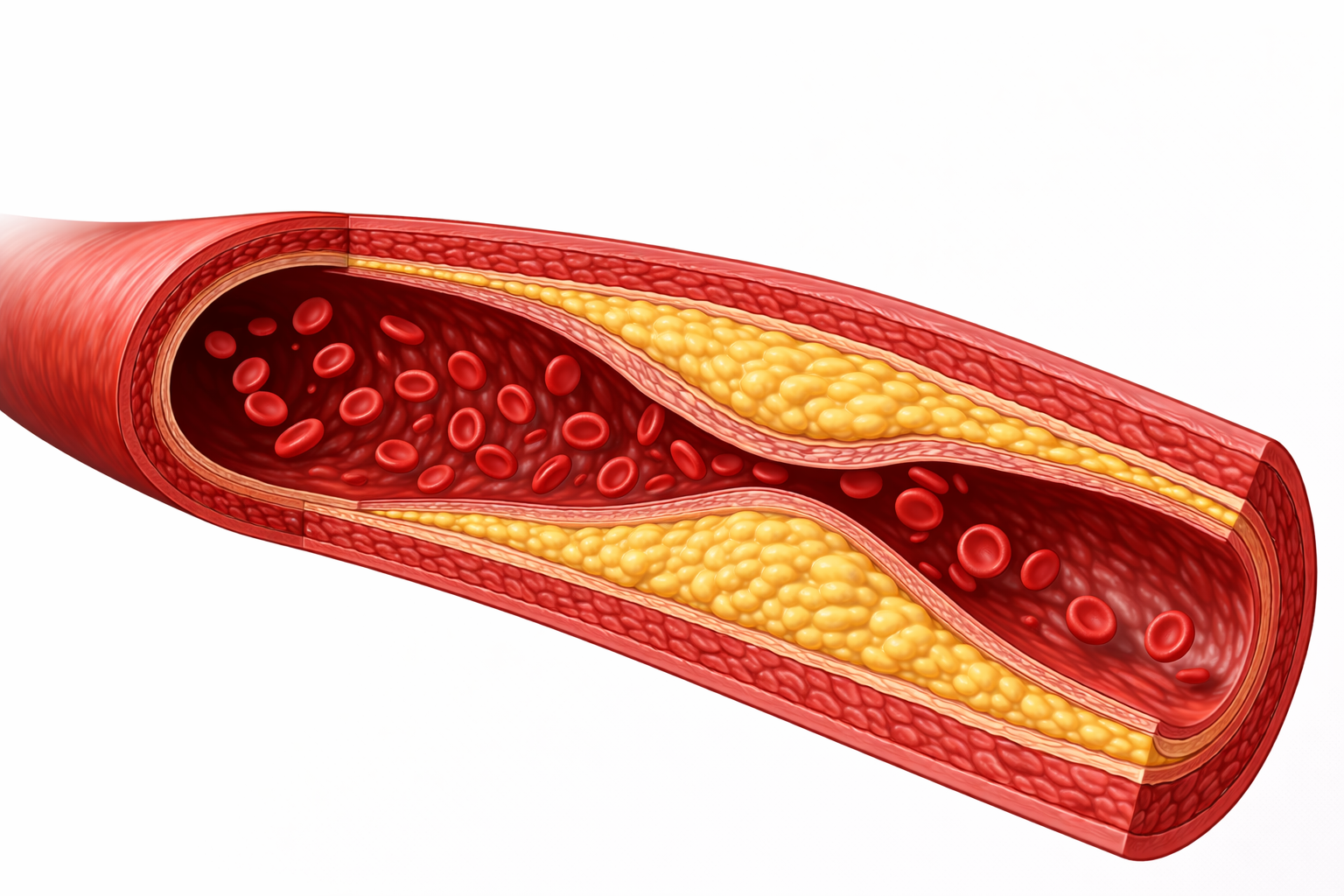
Image: Atherosclerotic plaque forms on the tunica intima, the innermost layer of the vessel wall. Lipid accumulation and inflammation occur beneath the endothelium, causing the plaque to enlarge within the intima and protrude into the lumen.
Beyond the Basics
Endothelial Dysfunction as the Initiating Event
The healthy endothelium plays an active regulatory role in vascular homeostasis. It promotes vasodilation through nitric oxide release, inhibits platelet adhesion, suppresses inflammation, and limits excessive lipid entry into the arterial wall. Exposure to cardiovascular risk factors such as hypertension, cigarette smoking, diabetes, dyslipidaemia, and chronic systemic inflammation disrupts these protective functions. Mechanical shear stress and biochemical injury reduce nitric oxide bioavailability, increase oxidative stress, and upregulate endothelial adhesion molecules such as VCAM-1 and ICAM-1. At the same time, endothelial permeability increases, allowing greater entry of circulating LDL particles into the intima.
As these changes accumulate, the endothelium shifts from a protective interface to a pro-inflammatory, pro-atherogenic surface. Leukocyte adhesion and migration are facilitated, lipid retention is enhanced, and local inflammatory signalling is amplified. This dysfunctional state sets the stage for plaque initiation and determines which arterial regions are most vulnerable to atherosclerotic change.
Lipid Infiltration, Oxidation, and Foam Cell Formation
Once LDL particles enter the intima, they become trapped and exposed to reactive oxygen species generated by endothelial cells and resident immune cells. Oxidation transforms LDL into a highly pro-inflammatory molecule that is toxic to endothelial cells and strongly chemotactic for circulating monocytes. Monocytes migrate into the arterial wall, differentiate into macrophages, and engulf oxidised LDL through scavenger receptors.
As lipid uptake continues, macrophages become lipid-laden foam cells. These cells accumulate within the intima and form fatty streaks, the earliest visible lesions of atherosclerosis. Fatty streaks can be detected even in childhood and adolescence. While not all fatty streaks progress to advanced plaques, their presence reflects ongoing endothelial dysfunction and local inflammation that may evolve further in the presence of persistent risk factors.
Plaque Maturation and Fibrous Cap Formation
With continued inflammatory signalling, smooth muscle cells migrate from the media into the intima. These cells proliferate and begin producing extracellular matrix components, particularly collagen. They may also ingest modified lipids, contributing to plaque growth. The result is formation of a fibrous cap that overlies a lipid-rich necrotic core composed of cholesterol crystals, dead foam cells, and cellular debris.
Plaque behaviour is determined more by composition than by size. Plaques with thick, collagen-rich fibrous caps and smaller lipid cores tend to remain stable. These lesions cause gradual luminal narrowing and predictable exertional symptoms due to fixed flow limitation. In contrast, plaques with thin fibrous caps, high inflammatory cell content, and large lipid cores are mechanically fragile. Ongoing inflammation weakens the cap, increasing susceptibility to rupture even when the degree of stenosis is only moderate.
The Vulnerable Plaque and Rupture Cascade
When mechanical stress and inflammation overwhelm fibrous cap integrity (how intact and stable the fibrous cap over an atherosclerotic plaque is), rupture occurs and triggers an acute thrombotic response. This sequence typically unfolds as:
Exposure of subendothelial collagen and tissue factor to circulating blood
Rapid platelet adhesion, activation, and aggregation
Activation of the coagulation cascade with fibrin deposition
Formation of an intraluminal thrombus that partially or completely occludes the artery
Complete occlusion of a coronary artery produces ST-elevation myocardial infarction, while partial or intermittent occlusion leads to non-ST-elevation myocardial infarction or unstable angina. Importantly, plaques that rupture are often not the most severely stenotic lesions beforehand. This explains why major cardiac events frequently arise from previously unrecognised disease rather than from long-standing, high-grade narrowing alone.
Consequences for Coronary Blood Flow and Myocardial Function
Atherosclerosis reduces myocardial perfusion through a combination of structural and functional changes in the coronary circulation. As plaques build up, they narrow the vessel lumen, which limits how much blood can flow through, particularly during periods of increased demand such as exercise. At the same time, endothelial dysfunction impairs the ability of the vessel to dilate (vasodilate) when more oxygen is needed. In more advanced disease, dysfunction of the small intramyocardial vessels (the microcirculation) further reduces effective blood delivery to the myocardium. When oxygen supply can no longer meet demand, myocardial ischaemia develops.
Ischaemia may present as typical angina, but it can also be silent, particularly in individuals with diabetes where pain perception is altered. Repeated or prolonged episodes of ischaemia begin to affect myocardial function. Contractility becomes impaired, the risk of arrhythmias increases due to electrical instability, and regions of myocardium may enter a state of stunning (temporary loss of contractile function after ischaemia) or hibernation (chronic reduction in function due to persistently reduced blood flow). Over time, these changes contribute to adverse ventricular remodelling, where the shape and function of the ventricle progressively deteriorate, leading to heart failure.
Progression to Coronary Artery Disease as a Clinical Spectrum
Coronary artery disease develops due to the progressive build-up of atherosclerotic plaques within the coronary arteries. These plaques can behave in different ways. Some remain stable and gradually narrow the vessel, limiting blood flow during increased demand and causing predictable exertional angina. Others are unstable and prone to rupture, which exposes underlying material and triggers clot formation, leading to acute coronary syndromes such as myocardial infarction.
Over time, repeated episodes of reduced blood flow can cause cumulative damage to the myocardium. This may occur as ongoing low-level ischaemia or through repeated acute events. As more myocardial tissue becomes impaired or non-functional, the left ventricle becomes less effective at pumping, eventually leading to chronic ischaemic cardiomyopathy, characterised by reduced contractility and diminished cardiac reserve.
Coronary artery disease is therefore not a single static condition but a spectrum of processes. It is driven by endothelial dysfunction (impaired regulation of the vessel lining), plaque formation and progression, inflammatory activity within the arterial wall, and thrombosis. At any point in time, the clinical presentation depends on which of these processes is dominant and how well the myocardium is able to compensate for reduced blood supply.
Clinical Connections
Atherosclerosis underpins the majority of cardiovascular morbidity and mortality, which is why long-term management focuses on modifying the processes that drive plaque growth and instability rather than treating luminal narrowing alone. Interventions such as smoking cessation, blood pressure control, glycaemic optimisation, and lipid-lowering therapy directly target endothelial dysfunction, inflammation, and lipid accumulation within the arterial wall. By reducing ongoing vascular injury, these strategies slow plaque progression and reduce the likelihood of rupture, even when established disease is already present.
Pharmacological therapy is particularly effective because it alters plaque biology rather than simply lowering risk markers. Statins reduce circulating LDL levels, but they also exert anti-inflammatory effects within the arterial wall and promote thickening of the fibrous cap, making plaques less prone to rupture. Blood pressure control reduces mechanical stress on the endothelium, while diabetes management limits glycation, oxidative stress, and microvascular damage. Together, these interventions reduce both chronic ischaemic burden and the risk of acute thrombotic events.
Certain symptom patterns should prompt concern for plaque instability or acute coronary compromise:
New-onset or rapidly worsening chest discomfort, especially with exertion or emotional stress
Chest pain associated with diaphoresis, nausea, or shortness of breath, suggesting acute ischaemia
Pain radiating to the arm, jaw, neck, or back, particularly when accompanied by haemodynamic instability
Understanding the pathophysiology of atherosclerosis supports early recognition of acute coronary syndromes, where plaque rupture and thrombosis abruptly limit myocardial blood flow. Early identification and escalation are critical because myocardial injury progresses rapidly once perfusion is compromised. Even outside acute settings, recognising changes in symptom pattern is essential, as stable disease can transition unpredictably to unstable plaque behaviour, marking a shift from chronic coronary disease to an immediately life-threatening condition.
Concept Check
How does endothelial dysfunction promote atherosclerosis?
Why is oxidised LDL more harmful than native LDL?
What differentiates a stable plaque from a vulnerable plaque?
Why can moderate stenosis still cause a myocardial infarction?
How does chronic myocardial ischaemia contribute to the development of heart failure?