MYOCARDIAL INFARCTION (STEMI & NSTEMI): The Pathophysiology of Acute Coronary Occlusion
An acute myocardial infarction (MI/AMI) occurs when blood flow to part of the heart is abruptly reduced or completely blocked, leading to ischaemia and irreversible necrosis of cardiac muscle. Most MIs result from rupture of an atherosclerotic plaque in a coronary artery, followed by thrombus formation that obstructs blood flow. MI is not a single event but a spectrum. In a ST-elevation MI (STEMI), complete occlusion of a coronary artery produces full-thickness (transmural) myocardial necrosis. In a non–ST-elevation MI (NSTEMI), partial or intermittent occlusion causes subendocardial necrosis without full-thickness involvement.
What You Need to Know
Myocardial infarction occurs when a coronary artery becomes acutely occluded, most often following rupture of an atherosclerotic plaque and superimposed thrombus formation. The sudden interruption of blood flow deprives downstream myocardium of oxygen and metabolic substrates. Within seconds of ischaemia, aerobic metabolism ceases and myocardial cells shift to anaerobic glycolysis. ATP stores fall rapidly, lactate accumulates, intracellular pH drops, and the contractile apparatus begins to fail. As a result, regional myocardial contraction is impaired almost immediately after occlusion.
If perfusion is not restored, cellular injury progresses in a time-dependent manner. Prolonged ischaemia disrupts cell membrane integrity, impairs ion pumps, and leads to calcium overload within cardiomyocytes, which accelerates cell death. The extent of myocardial injury is influenced by several factors, including the location of the affected artery, the size of the territory it supplies, the presence of collateral circulation, and the speed at which reperfusion is achieved. Earlier restoration of blood flow limits infarct size and preserves ventricular function.
Acute myocardial infarction can range from partial to complete coronary artery occlusion, with the degree of obstruction influencing the severity of myocardial injury.:
STEMI results from complete and sustained occlusion of a coronary artery, producing transmural myocardial necrosis and characteristic ST elevation on ECG.
NSTEMI occurs with partial occlusion or severe narrowing, causing subendocardial injury without ST elevation.
Both STEMI and NSTEMI involve irreversible myocardial injury and release of cardiac biomarkers, though the depth and extent of necrosis differ.
Despite differences in ECG appearance, both STEMI and NSTEMI represent acute coronary syndromes with significant risk of complications, including arrhythmias, heart failure, and cardiogenic shock. Rapid recognition and timely reperfusion or antithrombotic therapy are critical because myocardial damage progresses with each passing minute of ongoing ischaemia.
STEMI is diagnosed based on characteristic ECG changes in the appropriate clinical context and a blood test to identify cardiac troponins is used to confirm myocardial injury. Cardiac troponins are proteins released into the bloodstream when myocardial cells are injured. They are highly sensitive markers of myocardial injury but are not specific to the underlying cause, meaning they can be elevated in several types of myocardial infarction, as well as in other conditions such as myocarditis or severe sepsis. Diagnosis of myocardial infarction relies not just on a single elevated value, but on a dynamic rise and/or fall in troponin levels in the appropriate clinical context.
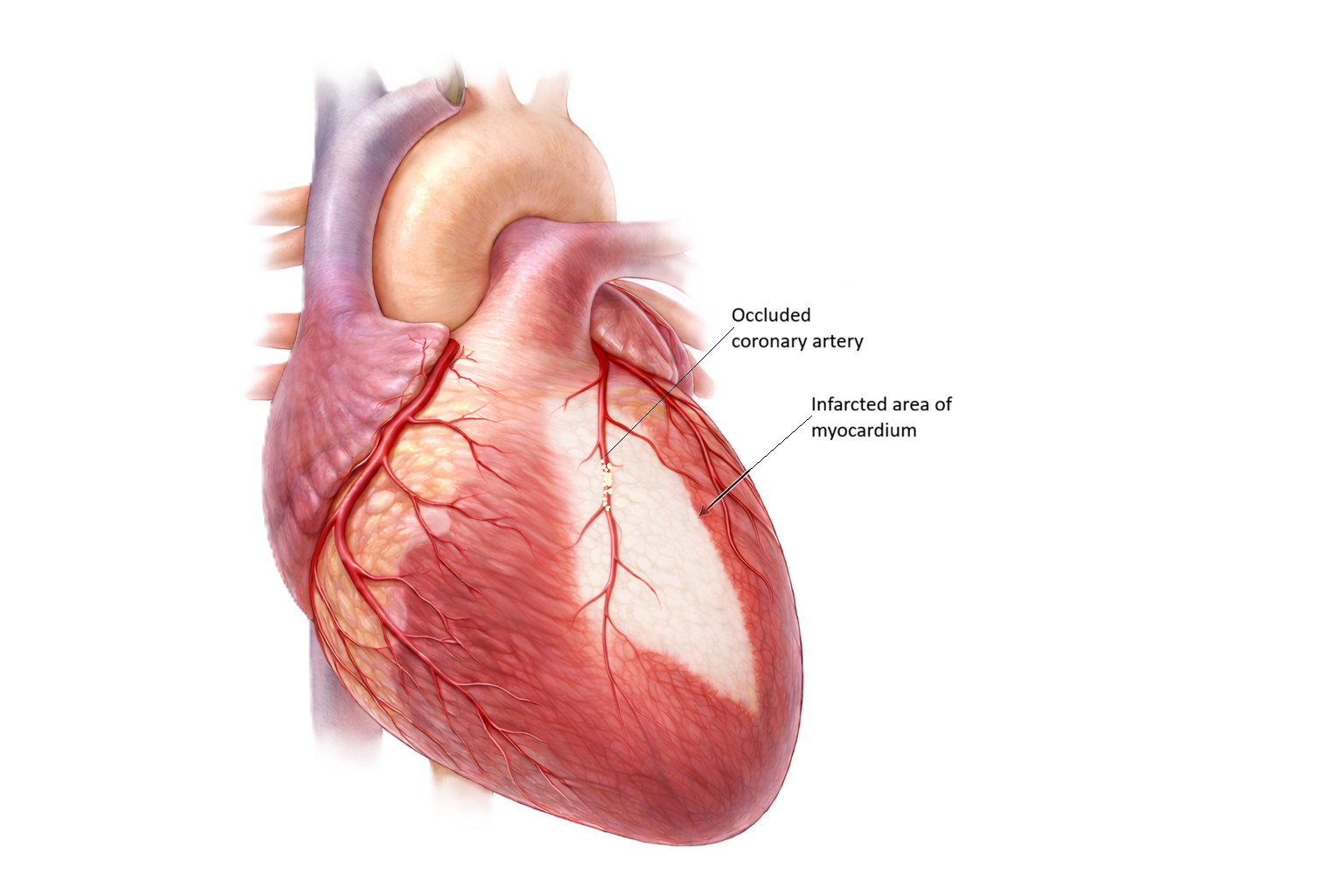
Image: The pale region represents infarcted myocardium, where prolonged interruption of coronary blood flow has led to irreversible tissue injury and loss of viable cardiac muscle.
Beyond the Basics
Type 1 vs Type 2 Myocardial Infarction
Type 1 and type 2 myocardial infarction differ in their underlying mechanism. Type 1 myocardial infarction is caused by an acute atherothrombotic event, usually plaque rupture or erosion within a coronary artery, which exposes thrombogenic material and leads to clot formation. This results in a sudden reduction or complete occlusion of coronary blood flow and causes myocardial necrosis.
In contrast, type 2 myocardial infarction occurs due to an imbalance between myocardial oxygen supply and demand in the absence of acute plaque rupture. This can occur in situations such as tachyarrhythmias, severe anaemia, hypoxia, or hypotension, where oxygen delivery is reduced or demand is increased. While coronary artery disease may still be present, the primary issue is not an acute thrombotic occlusion but a mismatch between supply and demand leading to ischaemic injury.
Plaque Rupture and Thrombus Formation
The initiating event in most myocardial infarctions is disruption of an atherosclerotic plaque within a coronary artery. Plaques that are rich in lipid and covered by a thin fibrous cap are mechanically fragile and prone to rupture or surface erosion. When this protective cap breaks, highly thrombogenic material within the plaque is suddenly exposed to circulating blood. Subendothelial collagen and tissue factor trigger rapid platelet adhesion and activation, followed by aggregation and activation of the coagulation cascade. Fibrin is deposited, stabilising the growing clot and producing a thrombus that obstructs coronary blood flow.
The degree of obstruction determines the clinical pattern. In ST-elevation myocardial infarction, thrombus formation produces complete and sustained occlusion of the artery, abruptly stopping blood flow to the downstream myocardium. In non–ST-elevation myocardial infarction, the thrombus only partially occludes the vessel or causes intermittent obstruction, allowing some residual flow but still producing significant myocardial injury.
Cellular and Metabolic Events During Myocardial Ischaemia
Once coronary blood flow is interrupted, myocardial cells experience immediate metabolic stress. Within seconds, oxygen delivery falls below the level required to support aerobic metabolism. Mitochondrial oxidative phosphorylation ceases, ATP production declines sharply, and contractile function begins to fail almost immediately. This early loss of contractility explains why regional wall motion abnormalities can be detected very soon after occlusion, sometimes even before pain is perceived.
As ischaemia persists, failure of energy-dependent ion pumps allows sodium and calcium to accumulate inside cardiomyocytes. Water follows sodium, leading to cellular swelling. Myocytes switch to anaerobic glycolysis to generate small amounts of ATP, but this produces lactate and hydrogen ions, lowering intracellular pH. The acidic environment and accumulation of metabolic by-products stimulate nociceptors, producing the characteristic chest pain of angina.
If blood flow is not restored, irreversible injury develops. The subendocardium, the inner layer of the myocardium, is affected first because it is furthest from the epicardial coronary blood supply and exposed to the highest wall stress. Cell membranes lose integrity, calcium overload accelerates mitochondrial damage, and cardiomyocytes undergo necrosis. With ongoing occlusion, this wave of cell death progresses outward toward the epicardium. In complete occlusion, this process culminates in full-thickness, or transmural, necrosis.
Why STEMI and NSTEMI Differ
The distinction between STEMI and NSTEMI reflects differences in the extent and depth of myocardial injury rather than the presence or absence of infarction. In STEMI, complete coronary occlusion produces transmural ischaemia that alters electrical currents across the full thickness of the ventricular wall. This generates ST-segment elevation on the ECG and is associated with a high risk of acute complications such as malignant arrhythmias, cardiogenic shock, and mechanical failure. Because ongoing occlusion rapidly expands infarct size, immediate reperfusion, ideally with percutaneous coronary intervention, is critical.
In NSTEMI, residual blood flow limits injury to the subendocardial region. ECG changes are usually limited to ST depression or T-wave inversion rather than ST elevation, but myocardial necrosis still occurs, as evidenced by elevated troponin levels. Although infarct size is typically smaller, the risk of recurrent ischaemia and subsequent infarction remains significant. Urgent treatment is required to stabilise the plaque, reduce thrombosis, and prevent progression, even if immediate reperfusion is not always necessary.
Reperfusion
The primary goal of acute myocardial infarction treatment is to restore blood flow to the affected area of myocardium as quickly as possible. This process is known as reperfusion. During an acute myocardial infarction, a coronary artery becomes partially or completely blocked, reducing oxygen delivery to cardiac muscle. The longer the obstruction remains, the greater the amount of myocardial damage that occurs.
Reperfusion aims to reopen the blocked artery and restore oxygenated blood flow before irreversible injury develops. This may be achieved through percutaneous coronary intervention (PCI), where a catheter is used to open the artery and often insert a stent, or through thrombolytic therapy, which uses medications to dissolve the clot. PCI is generally the preferred reperfusion strategy when it can be performed promptly.
Rapid reperfusion is associated with improved survival, reduced infarct size, preservation of cardiac function, and a lower risk of complications such as heart failure and cardiogenic shock. This is why early recognition of acute myocardial infarction and timely access to definitive treatment are critical components of care.
Reperfusion Injury
Restoring coronary blood flow is essential to salvage viable myocardium, but reperfusion itself can paradoxically contribute to further injury. Sudden reintroduction of oxygen generates reactive oxygen species that damage cell membranes and mitochondria. Rapid calcium influx worsens calcium overload, and inflammatory pathways are activated as neutrophils enter the reperfused tissue. These processes can provoke arrhythmias and transient myocardial dysfunction. Despite these effects, early reperfusion remains the most effective strategy to limit infarct size and improve survival, as the damage prevented by restoring flow far outweighs the injury caused by reperfusion.
Structural and Functional Consequences of Infarction
Following myocardial infarction, the affected region undergoes structural and functional changes that influence long-term outcome. Reperfused but injured myocardium may exhibit stunned myocardium, where contractility is temporarily depressed despite restored blood flow, gradually recovering over days to weeks. Chronically underperfused myocardium may enter a hibernating state, reducing contractile function to match limited oxygen supply while remaining viable.
Necrotic myocardium is ultimately replaced by fibrous scar tissue, which lacks contractile ability. To compensate, surviving myocardium undergoes hypertrophy and geometric remodelling. While initially adaptive, this remodelling increases wall stress and can lead to progressive systolic dysfunction and heart failure. Scar tissue also disrupts normal electrical conduction pathways, creating substrates for re-entry and increasing the risk of ventricular arrhythmias. Together, these changes explain why myocardial infarction is not a single event but the beginning of a chronic process that shapes long-term cardiac function and prognosis.
Clinical Connections
Myocardial infarction often presents with central chest pain described as pressure, tightness, or heaviness, frequently accompanied by diaphoresis, nausea, and pain radiating to the left arm, neck, or jaw. These symptoms result from acute myocardial ischaemia and activation of autonomic and inflammatory pathways. However, presentation is not uniform. Older adults, women, and individuals with diabetes may present without classic chest pain, instead reporting breathlessness, profound fatigue, epigastric discomfort, nausea, or syncope. These atypical patterns contribute to delayed recognition and highlight the need for a high index of suspicion when symptoms are unexplained or disproportionate to findings.
Early investigation is central to diagnosis and risk stratification. A 12-lead ECG should be obtained promptly to identify ST-segment elevation or ischaemic changes, such as ST depression or T-wave inversion. Cardiac troponins are proteins found in the myocardial cells, which are released into the bloodstream after myocardial injury or infarction. Blood tests that show elevated troponin levels confirm myocardial necrosis and help distinguish unstable angina from NSTEMI when ECG findings are nondiagnostic. Repeated ECGs and serial (taking the blood test at set intervals) troponin measurements are often required, as early changes may be subtle or evolve over time.
Key clinical priorities in the acute phase include:
Rapid identification of STEMI, where immediate reperfusion is required to limit infarct size
Recognition of evolving ischaemia or instability, such as recurrent chest pain, hypotension, or arrhythmias
Early detection of complications, including acute heart failure, cardiogenic shock, and malignant ventricular arrhythmias
Treatment strategies directly target the underlying pathophysiology. Reperfusion therapy restores coronary blood flow and limits myocardial necrosis. Antiplatelet agents and anticoagulants reduce thrombus propagation, beta blockers lower myocardial oxygen demand, nitrates improve coronary perfusion and relieve ischaemia, and statins stabilise plaques while reducing inflammation.
Ongoing assessment during hospitalisation and recovery focuses on detecting reinfarction, monitoring ventricular function, identifying arrhythmias, and supporting secondary prevention. Linking clinical findings to the mechanisms of ischaemia and infarction allows timely escalation, accurate interpretation of ECG changes, and anticipation of complications across both the acute and recovery phases.
Concept Check
What usually triggers the abrupt loss of coronary blood flow that causes MI?
Why is the subendocardium the first region to undergo irreversible necrosis?
What key pathological difference separates STEMI from NSTEMI?
How does anaerobic metabolism contribute to the chest pain experienced during an MI?
What are two long-term structural consequences of myocardial infarction?