CARDIOMYOPATHIES: Primary Disorders of Myocardial Structure and Function
Cardiomyopathies are a group of diseases characterised by structural and functional abnormalities of the myocardium that are not explained solely by coronary artery disease, valvular disorders or hypertension. These conditions impair the heart’s ability to generate effective contraction, relax appropriately or maintain normal chamber geometry, ultimately compromising cardiac output.
Cardiomyopathies may be inherited or acquired, and their clinical presentation ranges from asymptomatic disease to severe heart failure, arrhythmias and sudden cardiac death. Understanding the underlying pathophysiology is essential for recognising how changes in myocardial structure translate into impaired haemodynamics and clinical deterioration.
What You Need to Know
Cardiomyopathies are primary disorders of the myocardium that impair the heart’s ability to function as an effective pump. They are defined by abnormalities in myocardial structure, mechanics, or compliance rather than by valvular disease, hypertension, or congenital defects. Although causes vary, the defining feature is intrinsic dysfunction of the heart muscle itself.
Cardiomyopathies are classified according to the dominant structural and functional abnormality present:
Dilated cardiomyopathy, characterised by ventricular dilation and reduced systolic function
Hypertrophic cardiomyopathy, characterised by abnormal myocardial thickening and impaired ventricular filling
Restrictive cardiomyopathy, characterised by reduced ventricular compliance and impaired diastolic filling
Each subtype alters cardiac performance in a different way, but all interfere with the heart’s ability to maintain adequate cardiac output, particularly during increased physiological demand.
Structural changes in the myocardium disrupt normal ventricular geometry and pressure–volume relationships. These alterations affect stroke volume, ventricular filling pressures, and myocardial oxygen demand. As cardiac efficiency declines, intracardiac pressures rise and forward blood flow becomes increasingly limited.
In response to reduced cardiac output, compensatory neurohormonal mechanisms are activated. These include sympathetic nervous system stimulation and activation of the renin–angiotensin–aldosterone system. While initially supportive, these responses increase heart rate, afterload, and myocardial workload, promoting further remodelling and functional deterioration.
Over time, compensatory mechanisms become maladaptive. Progressive myocardial dysfunction increases the risk of heart failure, arrhythmias, thromboembolism, and sudden cardiac death. Symptoms often emerge late in the disease course, once compensatory capacity is exhausted, which explains why cardiomyopathies may remain clinically silent until significant dysfunction is present.
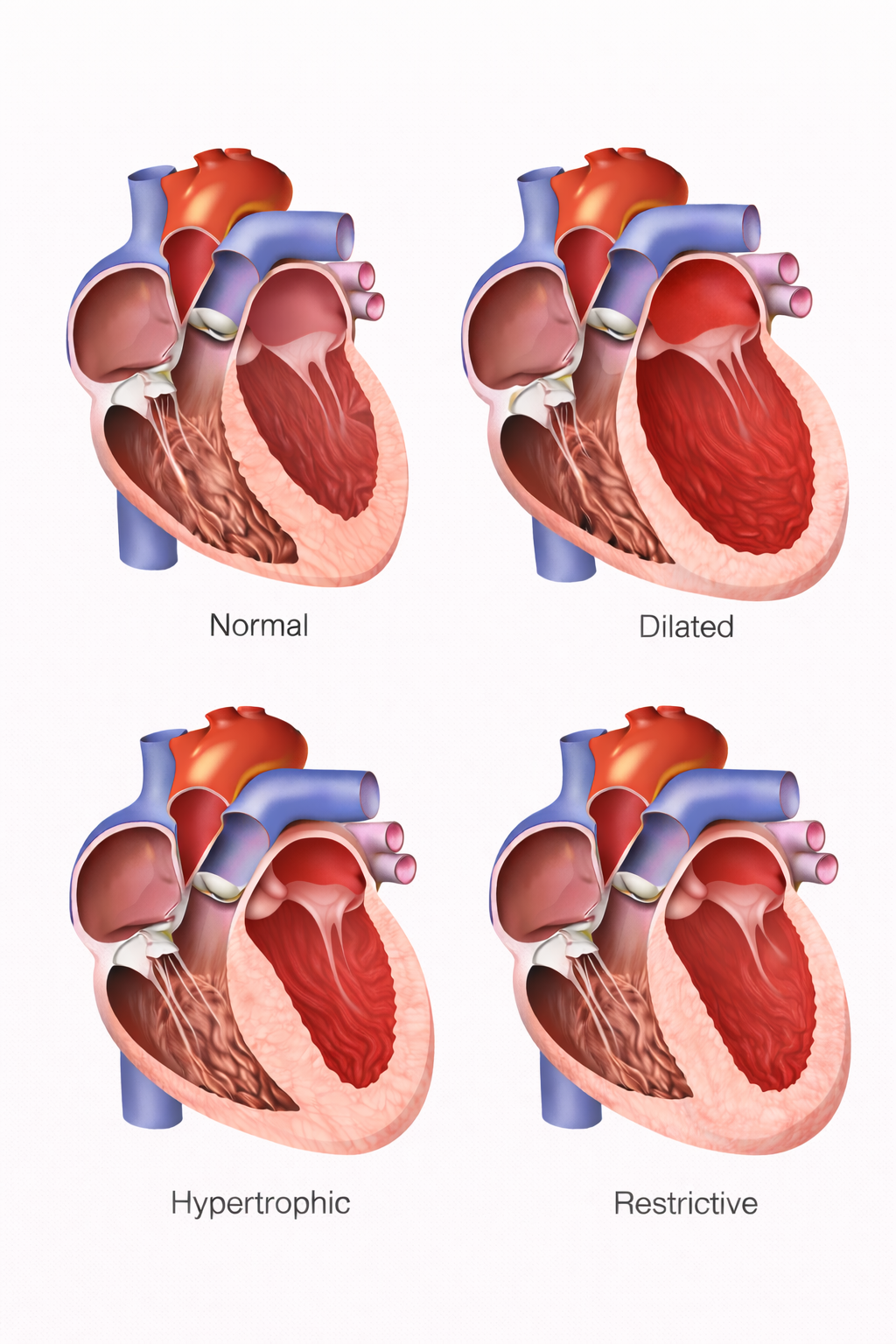
Image: Normal heart structure shows balanced chamber size and wall thickness with efficient function, while dilated cardiomyopathy is characterised by enlarged ventricles and reduced contractility, hypertrophic cardiomyopathy by thickened ventricular walls and a reduced chamber size, and restrictive cardiomyopathy by relatively normal-sized ventricles with stiff walls that impair filling.
Beyond the Basics
Dilated Cardiomyopathy: Systolic Failure and Ventricular Remodelling
Dilated cardiomyopathy is defined by ventricular chamber enlargement with impaired systolic function, most often affecting the left ventricle. As the ventricle dilates, myocardial fibres are stretched beyond their optimal length for contraction, reducing force generation and lowering ejection fraction. The ventricular wall may appear thin, but the key issue is loss of effective contractile performance rather than true wall thinning alone. Increased chamber radius raises wall stress according to the law of Laplace, meaning the myocardium must generate greater tension to eject blood, further worsening systolic dysfunction.
Multiple aetiologies can lead to this structural phenotype. Genetic mutations affecting cytoskeletal or sarcomeric proteins weaken force transmission during contraction, while acquired causes such as viral myocarditis result in myocyte injury and replacement fibrosis. Chronic alcohol exposure and cardiotoxic agents, including certain chemotherapeutic drugs, directly impair myocardial metabolism and contractility. Long-standing pressure or volume overload, such as that caused by uncontrolled hypertension or valvular regurgitation, can also drive progressive dilation when compensatory mechanisms fail.
Reduced forward flow triggers activation of the sympathetic nervous system and the renin–angiotensin–aldosterone system. These responses increase heart rate, vasoconstriction, and sodium and water retention, initially supporting perfusion but ultimately increasing afterload and ventricular filling pressures. Over time, this neurohormonal activation accelerates adverse remodelling, promotes further dilation, and contributes to the transition from compensated dysfunction to overt heart failure.
Hypertrophic Cardiomyopathy: Diastolic Dysfunction and Dynamic Obstruction
Hypertrophic cardiomyopathy is most commonly an inherited disorder caused by mutations in sarcomeric proteins such as beta-myosin heavy chain or myosin-binding protein C. These mutations lead to abnormal myocyte growth and disorganised fibre arrangement, known as myocyte disarray, resulting in inappropriate myocardial thickening that is not explained by loading conditions. Hypertrophy most often involves the interventricular septum, though patterns can vary.
The dominant functional problem in hypertrophic cardiomyopathy is impaired diastolic filling. The thickened myocardium becomes stiff and poorly compliant, meaning higher filling pressures are required to achieve adequate ventricular filling during diastole. Although systolic contraction is usually preserved or even hyperdynamic, reduced end-diastolic volume limits stroke volume, particularly during exercise when diastolic filling time shortens. In addition, myocardial hypertrophy increases oxygen demand while simultaneously compressing intramural coronary vessels, predisposing to myocardial ischaemia even in the absence of coronary artery disease.
In some individuals, septal hypertrophy narrows the left ventricular outflow tract during systole, creating dynamic obstruction. This obstruction is often exacerbated by reduced preload or increased contractility, such as during exertion or dehydration. The resulting pressure gradient can cause exertional dyspnoea, chest pain, syncope, and increases the risk of malignant ventricular arrhythmias and sudden cardiac death, particularly in younger patients.
Restrictive Cardiomyopathy: Loss of Ventricular Compliance
Restrictive cardiomyopathy is characterised by markedly reduced ventricular compliance with relatively preserved systolic function, at least in early disease. Unlike hypertrophic cardiomyopathy, ventricular wall thickness may appear normal, but the myocardium is functionally stiff and unable to relax appropriately during diastole. This impaired relaxation limits ventricular filling and leads to elevated diastolic pressures.
The underlying mechanism is most often infiltration or fibrosis of the myocardium. Conditions such as amyloidosis involve deposition of abnormal proteins within the myocardial interstitium, while sarcoidosis produces granulomatous inflammation that disrupts normal tissue architecture. Haemochromatosis causes iron accumulation within cardiomyocytes, interfering with cellular function, and prior chest radiation can induce diffuse myocardial fibrosis. Regardless of cause, the end result is a ventricle that resists expansion despite normal contractile strength.
Elevated filling pressures are transmitted backward into the atria and venous circulation, leading to biatrial enlargement and prominent signs of systemic and pulmonary congestion. Stroke volume remains limited because ventricular filling cannot increase, particularly during exertion, contributing to exercise intolerance and fatigue despite a preserved ejection fraction.
Electrical Instability and Arrhythmogenesis
Structural abnormalities in cardiomyopathies disrupt normal electrical conduction through the myocardium. Fibrosis, hypertrophy, and chamber dilation create areas of delayed conduction and heterogeneous refractoriness, which favour re-entry circuits and ectopic activity. These changes increase susceptibility to both atrial arrhythmias, such as atrial fibrillation, and ventricular arrhythmias.
In hypertrophic cardiomyopathy, myocardial disarray and fibrosis contribute to electrical instability independent of systolic function. In dilated cardiomyopathy, chamber enlargement and scarring from prior inflammation or injury increase the risk of ventricular tachyarrhythmias as disease advances. Importantly, arrhythmic risk does not always correlate with symptom severity or degree of heart failure, which is why rhythm monitoring and risk stratification are central to management.
Progression to Heart Failure
Although the initial pathophysiology differs, all cardiomyopathies share a tendency toward progressive impairment of cardiac output. Reduced forward flow leads to fatigue, weakness, and organ hypoperfusion, while elevated filling pressures cause pulmonary congestion and peripheral oedema. Neurohormonal activation, while initially adaptive, increases myocardial workload and promotes further remodelling and fibrosis over time.
As structural and functional abnormalities worsen, compensatory mechanisms become maladaptive, and patients may progress to advanced heart failure. At this stage, pharmacological therapy may be insufficient, and some individuals require advanced interventions such as mechanical circulatory support or cardiac transplantation.
Clinical Connections
Cardiomyopathies often come to clinical attention through symptoms that overlap with other cardiovascular conditions, which is why pattern recognition matters. Breathlessness and fatigue typically reflect reduced forward flow or elevated filling pressures, while palpitations may signal atrial or ventricular arrhythmias arising from structurally abnormal myocardium. Syncope, particularly during exertion, should raise suspicion for hypertrophic cardiomyopathy with dynamic outflow obstruction or malignant ventricular arrhythmia. Some individuals remain asymptomatic and are diagnosed incidentally during imaging for unrelated reasons, whereas others present acutely with pulmonary oedema, sustained ventricular tachycardia, or sudden cardiac arrest.
Although presentation may appear similar across subtypes, the underlying physiology differs and influences assessment priorities. Key distinctions include:
Dilated cardiomyopathy: progressive exertional dyspnoea, orthopnoea and peripheral oedema linked to reduced ejection fraction and chamber enlargement.
Hypertrophic cardiomyopathy: exertional chest pain, presyncope or syncope due to diastolic dysfunction and possible outflow tract obstruction, sometimes with a family history of sudden death.
Restrictive cardiomyopathy: prominent systemic congestion, ascites or peripheral oedema with relatively preserved systolic function but markedly impaired filling.
Echocardiography is used to diagnosis because it visualises chamber size, wall thickness, systolic performance, and diastolic filling patterns in real time. In dilated cardiomyopathy, imaging typically demonstrates ventricular enlargement with reduced ejection fraction. In hypertrophic cardiomyopathy, asymmetric septal thickening and abnormal diastolic relaxation are characteristic, and dynamic outflow gradients may be provoked. Restrictive cardiomyopathy often shows normal ventricular size with biatrial enlargement and restrictive filling patterns.
Cardiac magnetic resonance imaging adds further structural detail, identifying areas of fibrosis through late gadolinium enhancement and detecting infiltrative processes such as amyloidosis. Management is tailored to the underlying type and risk profile, ranging from guideline-directed heart failure pharmacotherapy to rhythm control strategies, implantable cardioverter-defibrillators for sudden death prevention, or advanced therapies including transplantation in end-stage disease.
Concept Check
How does ventricular dilation impair systolic function in dilated cardiomyopathy?
Why does hypertrophic cardiomyopathy primarily affect diastolic filling?
What structural changes limit ventricular compliance in restrictive cardiomyopathy?
Why are cardiomyopathies strongly associated with arrhythmias?
How does neurohormonal activation contribute to disease progression across all cardiomyopathies?