WOUND HEALING — Stage 1: Haemostasis
Haemostasis is the first and fastest stage of wound healing, beginning the instant blood vessels are disrupted. Although often summarised simply as “blood clotting,” haemostasis is in reality a highly coordinated cellular and biochemical event that establishes the entire foundation of wound repair. Within seconds of injury, the body must accomplish multiple tasks simultaneously: prevent life-threatening blood loss, seal exposed tissues from the external environment, and initiate a cascade of intracellular signalling that dictates the inflammatory and proliferative responses to follow. The haemostatic response is therefore both mechanical and molecular, providing an immediate physical barrier to bleeding and a biochemical scaffold that programs later healing events.
What You Need to Know
Haemostasis is the first and most immediate phase of wound healing, beginning within seconds of tissue injury. Its primary purpose is to stop blood loss while creating the initial structural and biochemical foundation for repair. This phase relies on rapid vascular responses, platelet activation and coagulation factor cascades that work together to seal damaged vessels and stabilise the wound environment.
Key processes involved in haemostasis include:
Reflex vasoconstriction that limits blood flow at the injury site
Platelet adhesion and aggregation on exposed collagen and von Willebrand factor
Activation of the coagulation cascade leading to fibrin clot formation
Release of platelet-derived growth factors that signal subsequent healing phases
As platelets adhere and aggregate, they release mediators such as ADP, thromboxane A₂ and serotonin, which amplify platelet recruitment and strengthen the developing plug. Simultaneously, exposure of tissue factor activates the extrinsic coagulation pathway, generating thrombin and converting fibrinogen into fibrin. Fibrin strands interlace with platelets to form a stable clot that seals the wound and resists mechanical disruption. Importantly, this clot is not inert. It acts as a provisional extracellular matrix rich in growth factors such as PDGF, TGF-β and VEGF, which guide immune cell recruitment, angiogenesis and fibroblast migration. The effectiveness of haemostasis therefore directly influences how efficiently the wound transitions into inflammation and subsequent repair.
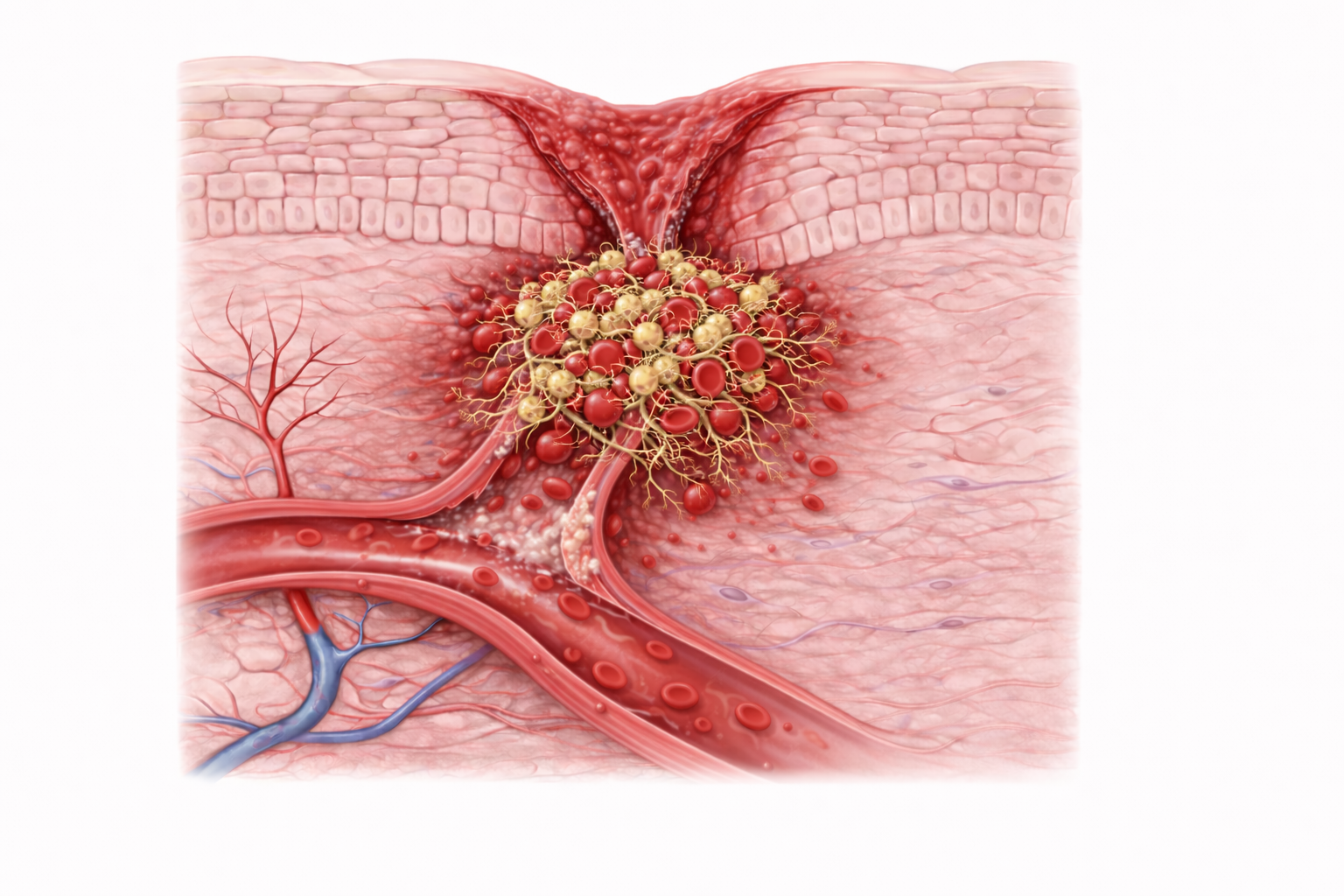
Image: Haemostasis begins immediately after injury, with vasoconstriction reducing blood flow and platelets adhering to exposed collagen at the damaged vessel wall. Activated platelets aggregate to form a temporary plug, while a fibrin mesh stabilises the clot and traps red blood cells, effectively sealing the wound and limiting further blood loss.
Beyond the Basics
Immediate vascular responses and endothelial signalling
Haemostasis begins with an immediate vasoconstrictive response at the site of injury. This response is driven by neural reflexes and by endothelin-1 released from damaged endothelial cells. Vasoconstriction rapidly reduces local blood flow, limiting blood loss and lowering shear forces within the vessel. This slowing of flow is essential, as it allows platelets and coagulation proteins to remain in close proximity to the injury site rather than being washed away by circulating blood.
Disruption of the endothelial lining simultaneously exposes the subendothelial matrix, which is rich in collagen, laminin, fibronectin and von Willebrand factor. Under normal conditions, intact endothelium maintains an anticoagulant surface. Injury abruptly reverses this balance, converting the local environment into a pro-coagulant state. Exposure of these matrix components provides the biochemical cues required for platelet adhesion and marks the true initiation of haemostasis.
Platelet adhesion, activation and aggregation
Platelets are the primary cellular mediators of early haemostasis. They adhere to exposed collagen through surface receptors such as glycoprotein Ia/IIa and bind von Willebrand factor via the GP Ib receptor. Adhesion triggers rapid platelet activation, during which platelets undergo dramatic morphological change, spreading across the damaged surface and extending pseudopodia to increase contact area.
Activated platelets release dense and alpha granules containing ADP, calcium, serotonin and thromboxane A₂. These mediators amplify platelet recruitment, reinforce vasoconstriction and promote further aggregation. ADP and thromboxane A₂ activate GP IIb/IIIa receptors, allowing fibrinogen to cross-link adjacent platelets into a cohesive plug. In parallel, platelets release growth factors including PDGF, TGF-β, IGF-1 and VEGF. These signals initiate inflammatory cell recruitment, angiogenic priming and fibroblast chemotaxis, positioning platelets as both mechanical stabilisers and early regulators of tissue repair.
Coagulation cascade and fibrin clot stabilisation
While platelets form a temporary plug, exposure of tissue factor at the wound site activates the extrinsic coagulation pathway. This rapidly generates thrombin, a central enzyme in haemostasis. Thrombin converts soluble fibrinogen into insoluble fibrin monomers, which polymerise into a stabilising mesh over and between platelets.
Thrombin also activates factor XIII, which cross-links fibrin strands, greatly increasing clot strength and resistance to mechanical disruption. Calcium ions released from activated platelets act as essential cofactors throughout the coagulation cascade, highlighting the tight functional integration between cellular and plasma components of haemostasis.
The resulting platelet–fibrin clot seals the wound and anchors firmly to surrounding tissue. Importantly, this clot also functions as a temporary extracellular matrix, supporting cell adhesion and migration while retaining cytokines, chemokines and growth factors required for later healing stages.
Vasoconstriction and the programmed transition to vasodilation
Although vasoconstriction is essential for effective haemostasis, it is deliberately short-lived. Once a stable clot has formed and bleeding is controlled, continued vasoconstriction would impair healing by limiting immune access, oxygen delivery and nutrient supply. At this point, the vascular response must shift toward vasodilation and increased permeability, signalling the transition from haemostasis to inflammation.
This transition is driven by inflammatory mediators released from platelets, damaged cells and activated endothelium, including histamine, prostaglandins, nitric oxide and bradykinin. These signals override earlier constrictive influences, causing blood vessels to dilate and endothelial junctions to loosen. Vasodilation increases blood flow to the wound and allows plasma proteins, neutrophils and monocytes to exit the circulation and enter the tissue. This change enables immune defence, supports early debridement and creates the moist, protein-rich environment necessary for cell migration.
The clot as a coordinating hub for subsequent healing
The haemostatic clot is not an inert barrier but an active signalling structure that coordinates the next phases of healing. Platelet-derived growth factors regulate leukocyte recruitment, endothelial proliferation and fibroblast activation. Transforming growth factor beta influences inflammatory resolution and later collagen synthesis, while vascular endothelial growth factor primes the wound bed for angiogenesis.
Through these biochemical signals, haemostasis establishes both structural stability and biological direction. Effective clot formation ensures that inflammation begins in a controlled, spatially organised manner, setting the conditions for efficient proliferation and remodelling. Failure or dysregulation at this stage compromises the entire healing cascade.
Clinical Connections
Effective haemostasis is essential for wound progression, as it determines whether healing can move efficiently into inflammation. When clot formation is delayed or unstable, bleeding persists and the wound environment remains disorganised, preventing immune cells and growth factors from acting in a coordinated way. Disorders that affect platelets, coagulation factors or endothelial integrity therefore have a disproportionate impact at this earliest stage of healing.
In clinical settings, haemostatic dysfunction commonly presents through the following patterns:
Prolonged bleeding and delayed inflammatory onset due to impaired clot formation
Fragile or easily disrupted clots that fail to act as a stable scaffold
Microvascular compromise where excessive clotting limits perfusion and oxygen delivery
Patients with thrombocytopenia, haemophilia or vitamin K deficiency may struggle to generate a stable fibrin network, while those taking anticoagulants such as warfarin, heparin or direct oral anticoagulants often experience prolonged oozing rather than effective clot consolidation. In these situations, inflammation is delayed and subsequent stages of healing are poorly synchronised. Conversely, excessive or dysregulated clotting can impair microcirculatory flow, particularly in already vulnerable tissue, leading to local hypoxia and compromised repair.
Systemic factors further modify haemostatic quality. Diabetes, ageing and smoking impair platelet responsiveness and endothelial signalling, weakening vasoconstriction, platelet adhesion and growth factor release. As a result, the early wound environment may lack the structural stability and biochemical cues required for effective progression.
Early assessment of clot quality provides valuable prognostic insight. A firm, well-adhered fibrin clot supports immune recruitment and healthy granulation tissue formation, whereas a weak, unstable or infected clot increases the risk of delayed healing, wound breakdown or chronic non-healing wounds. Recognising haemostatic failure early allows clinicians to adjust management strategies, address systemic contributors and reduce downstream complications.
Concept Check
Why is the exposure of subendothelial collagen a critical trigger for platelet activation?
How do platelets act as both structural components and signalling cells during haemostasis?
Explain the role of thrombin in stabilising the clot and preparing the wound for later healing.
Why might patients on anticoagulant therapy experience impaired early wound healing?
How does the fibrin clot influence inflammation, angiogenesis and fibroblast migration?