Acute Tubular Necrosis (ATN): Ischaemic and Toxic Tubular Injury in Acute Kidney Failure
Acute tubular necrosis is a common cause of intrinsic acute kidney injury characterised by direct injury and death of renal tubular epithelial cells. It most often develops following prolonged renal hypoperfusion or exposure to nephrotoxic substances and represents a transition from functional renal impairment to structural damage. The pathophysiology of ATN explains why kidney function fails to recover quickly after shock or toxin exposure, why urine findings change, and why recovery occurs in phases rather than immediately after the initial insult resolves.
What You Need to Know
Renal tubules are the workhorses of the nephron. They require large amounts of oxygen and energy to move electrolytes, water, and waste products between the blood and the forming urine. In acute tubular necrosis (ATN), these cells are injured by reduced blood flow (ischaemia) or by toxins such as drugs, contrast agents, or myoglobin, disrupting their ability to maintain normal transport and cellular structure.
When tubular cells are damaged, several key problems develop:
injured cells detach and clog the tubules, slowing or blocking urine flow
surviving cells cannot reabsorb water and electrolytes properly
waste products are no longer cleared efficiently from the blood
Even if kidney blood flow improves, these structural and functional changes mean the kidneys cannot immediately resume normal filtration and urine production. As a result, ATN often causes a period of reduced or ineffective kidney function before recovery can begin.
This explains why ATN is a common cause of acute kidney injury in critically ill patients and why recovery depends not just on restoring circulation, but on allowing time for the damaged tubules to repair and regenerate.
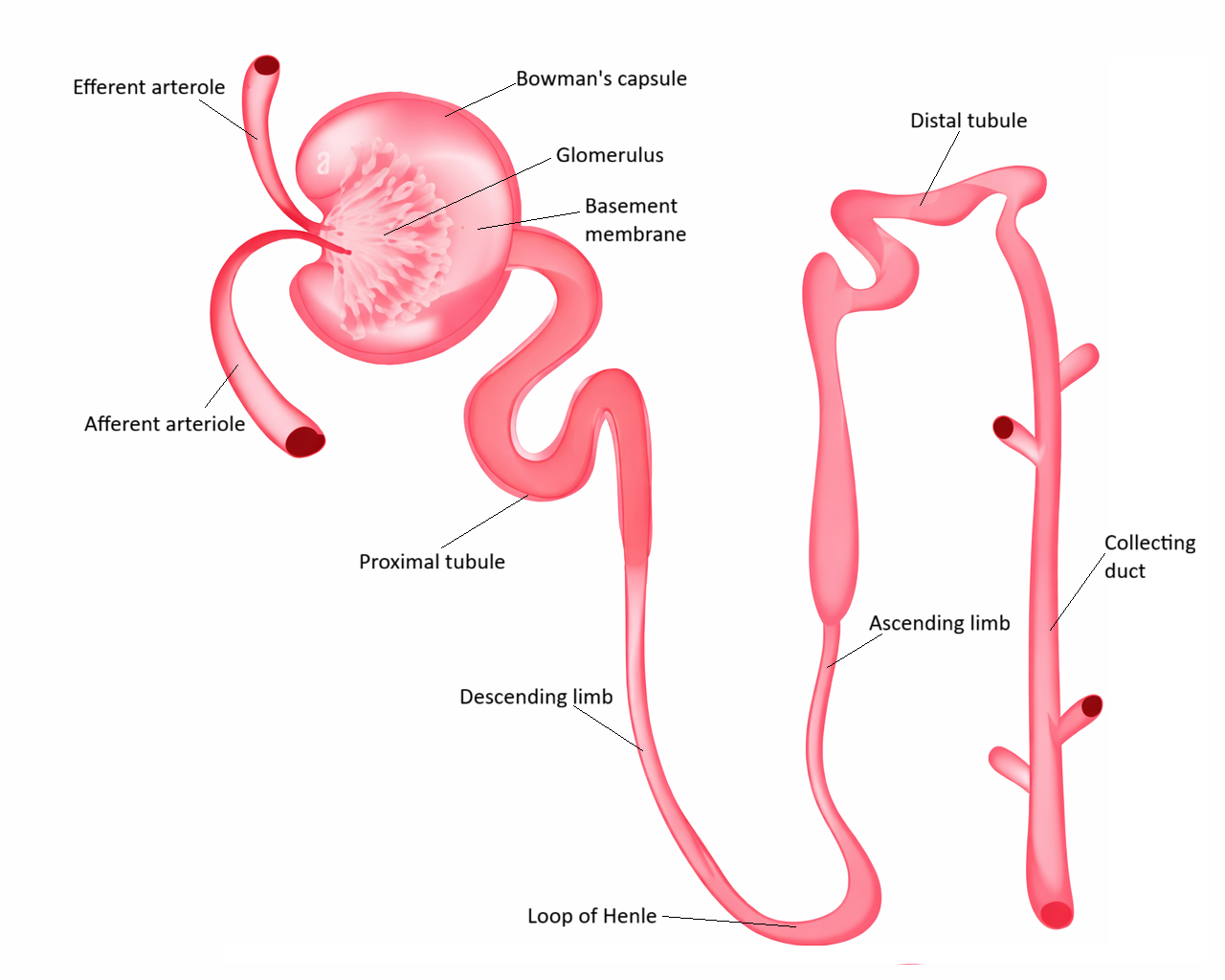
Image: Renal tubules are responsible for reabsorption and secretion, refining filtrate into urine. In acute tubular necrosis, injury to tubular epithelial cells disrupts these processes, leading to impaired reabsorption, tubular obstruction, and reduced kidney function.
Beyond the Basics
Ischaemic Injury and Oxygen Deprivation
Ischaemic ATN develops when renal perfusion falls below the level required to sustain tubular cell metabolism, as occurs during prolonged hypotension, septic shock, or major trauma. Although the kidney initially compensates through autoregulatory mechanisms that preserve glomerular blood flow, these defences cannot sustain oxygen delivery when hypoperfusion persists. Tubular epithelial cells, particularly in the proximal tubule and thick ascending limb, are especially vulnerable because they perform energy-intensive transport processes while operating in regions of relatively low oxygen tension.
As oxygen delivery falls, mitochondrial ATP production declines, impairing sodium-potassium pumps and other active transport systems. This leads to intracellular sodium and calcium accumulation, cellular swelling, and disruption of membrane integrity. Mitochondrial injury further amplifies oxidative stress, pushing the cell toward apoptosis or necrosis and compromising the structural integrity of the nephron.
Nephrotoxic Injury and Direct Cellular Damage
In toxic ATN, tubular injury occurs even when renal perfusion is preserved. Drugs, contrast agents, heavy metals, myoglobin, and endogenous toxins enter the tubular lumen and are taken up by epithelial cells, where they disrupt mitochondrial function, damage cell membranes, and generate reactive oxygen species. These effects interfere with energy production and destabilise the cytoskeleton, causing cells to detach from the basement membrane.
Because this injury is driven by direct cellular toxicity rather than hypoxia, ATN may develop in patients who appear haemodynamically stable. This explains why kidney injury can follow procedures, chemotherapy, or rhabdomyolysis even in the absence of overt shock or hypotension.
Tubular Obstruction and Back-Leak of Filtrate
As injured tubular epithelial cells die and detach, they combine with Tamm-Horsfall proteins to form granular casts that accumulate within the tubular lumen. These casts obstruct flow, increasing intratubular pressure and mechanically opposing filtration at the glomerulus. Even when blood flow improves, this obstruction limits urine formation.
At the same time, disruption of tight junctions between tubular cells allows filtrate to leak backward into the interstitium and circulation. This means that some plasma may still be filtered at the glomerulus, but little of it reaches the bladder as urine, because it is reabsorbed through damaged tubules rather than excreted.
Loss of Tubular Selectivity
Healthy renal tubules precisely control the movement of electrolytes and water, allowing sodium, bicarbonate, and water to be reabsorbed while potassium, hydrogen ions, and waste products are secreted. In ATN, this selectivity collapses because transporters are damaged and epithelial polarity is lost.
As a result, sodium reabsorption falls, urine becomes relatively dilute, and electrolyte abnormalities develop even if filtration is partially preserved. This physiological failure distinguishes ATN from pre-renal kidney injury, where tubular function remains intact and sodium conservation is preserved.
Inflammatory Amplification and Microvascular Dysfunction
Tubular injury does not remain confined to epithelial cells. Damaged cells release cytokines and danger signals that activate endothelial cells and recruit immune cells into the renal microcirculation. This leads to capillary congestion, interstitial oedema, and impaired oxygen delivery, even after systemic perfusion has been restored.
This inflammatory and microvascular dysfunction creates a vicious cycle in which hypoxia and inflammation sustain further tubular injury. ATN therefore behaves as an evolving disease process rather than a single moment of injury.
Phases of ATN and Renal Recovery
ATN progresses through recognisable physiological phases. During the initiation phase, hypoxia or toxicity produces early cellular injury and declining filtration. The maintenance phase is characterised by sustained reduction in glomerular filtration, oliguria, and accumulation of metabolic waste as tubular function remains impaired.
Recovery begins when surviving tubular cells proliferate, migrate, and re-establish a continuous epithelial lining along the basement membrane. Transport proteins are re-expressed and normal polarity gradually returns, allowing reabsorption and secretion to recover. The completeness of this repair depends on the severity and duration of injury, which is why some patients regain normal renal function while others progress to chronic kidney disease.
Clinical Connections
Acute tubular necrosis commonly presents with persistent oliguria, rising creatinine, electrolyte disturbance and metabolic acidosis that do not resolve with initial fluid resuscitation. Unlike pre-renal states, urine findings point to intrinsic tubular injury rather than adaptive sodium and water conservation. Damaged tubular cells lose the ability to reabsorb sodium and concentrate urine, leading to inappropriate urinary sodium loss, impaired acid excretion and accumulation of potassium and nitrogenous waste. These abnormalities reflect cellular dysfunction within the nephron rather than inadequate circulating volume alone.
Several clinical and laboratory features help distinguish ATN from haemodynamic causes of acute kidney injury:
Persistent oliguria or non-oliguria despite restoration of perfusion
Urinalysis showing granular or “muddy brown” casts, consistent with sloughed tubular cells
Fractional excretion of sodium that remains inappropriately elevated due to impaired tubular reabsorption
Diagnosis is based on the clinical context, time course and urine findings rather than a single test. ATN typically follows a clear insult such as prolonged hypotension, sepsis, major surgery, rhabdomyolysis or exposure to nephrotoxins. Serial monitoring of urine output, creatinine, electrolytes and acid–base status is essential because deterioration may continue during the maintenance phase even after the initial insult has resolved.
Management centres on limiting further tubular injury and supporting homeostasis while regeneration occurs. Perfusion must be maintained without precipitating fluid overload, nephrotoxic exposures must be avoided, and electrolyte and acid–base abnormalities corrected proactively. Renal replacement therapy is initiated when complications such as refractory hyperkalaemia, severe acidosis, fluid overload or uraemic symptoms develop. Dialysis supports physiological stability during the maintenance phase but does not hasten tubular repair, which depends on time, removal of the injurious stimulus and recovery of viable epithelial cells.
Concept Check
Why are renal tubular cells particularly vulnerable to ischaemic injury?
How does tubular obstruction reduce effective urine formation in ATN?
Why does ATN persist after renal perfusion is restored?
How does toxic ATN differ mechanistically from ischaemic ATN?
Why does recovery from ATN occur in phases rather than immediately?