The Neuromuscular Junction: Structure, Signal Transmission & Control of Contraction
The neuromuscular junction (NMJ) is the highly specialised synapse where motor neurons communicate with skeletal muscle fibres to initiate voluntary movement. Every conscious movement, from breathing and posture to fine hand control, depends on precise signal transmission across this microscopic interface. The NMJ converts an electrical signal in a nerve into a mechanical response in muscle through a tightly regulated sequence of chemical and electrical events. Because of its central role in movement, posture, respiration, and strength, disruption of NMJ function produces profound clinical consequences ranging from fatigable weakness to complete paralysis.
What You Need to Know
The neuromuscular junction (NMJ) is the specialised synapse between a motor neuron and a skeletal muscle fibre. It is the site where electrical signals in nerves are converted into chemical signals that trigger muscle contraction. Because every voluntary movement depends on this connection, the NMJ must operate with extreme speed and reliability to ensure that nerve impulses consistently produce the intended muscle response.
The neuromuscular junction consists of specialised structures that enable efficient transmission of signals from nerve to muscle, including:
The presynaptic motor neuron terminal contains synaptic vesicles filled with acetylcholine (ACh) and voltage-gated calcium channels
The synaptic cleft is the narrow space where ACh is released and diffuses toward the muscle
The postsynaptic muscle end plate contains densely packed nicotinic ACh receptors arranged in junctional folds that amplify the signal
ACh binding opens ion channels, causing depolarisation of the muscle membrane and initiation of an action potential
Motor neurons release acetylcholine only at the neuromuscular junction, and skeletal muscle fibres respond only to this neurotransmitter. This creates a highly specific and dependable link between nerve and muscle, ensuring that each nerve impulse leads to a predictable muscle contraction. This one-to-one signalling allows precise voluntary control of movement, from powerful limb actions to fine motor tasks such as writing or speech.
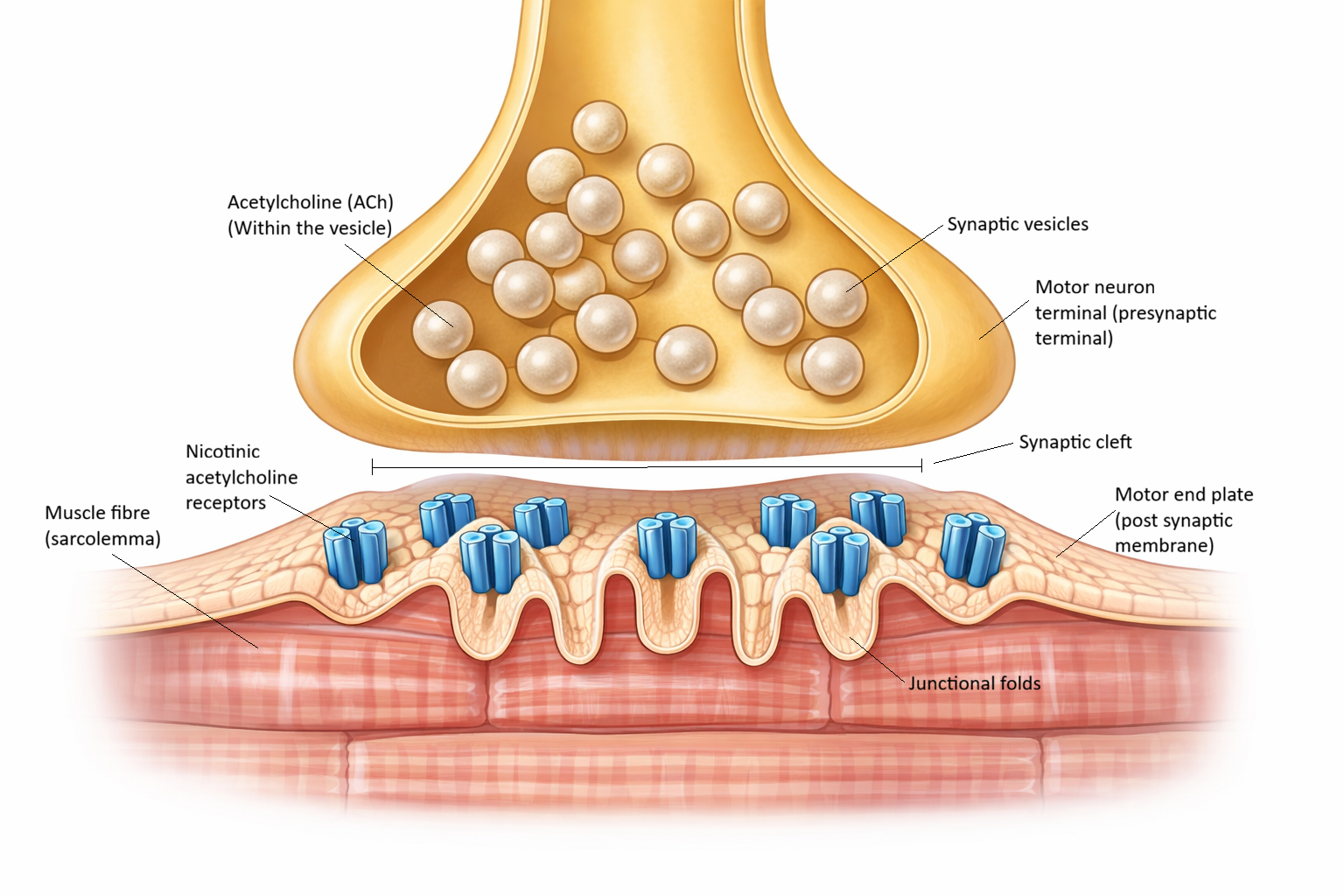
Image: Neuromuscular junction showing a motor neuron terminal releasing acetylcholine into the synaptic cleft, with junctional folds of the muscle end plate increasing surface area for receptors and enabling efficient transmission from nerve to muscle.
Beyond the Basics
Signal Transmission at the Neuromuscular Junction
When an action potential reaches the terminal of a motor neuron, the change in electrical charge opens voltage-gated calcium channels in the presynaptic membrane. Calcium ions rush into the terminal, acting as the critical trigger for neurotransmitter release. This influx causes synaptic vesicles filled with acetylcholine (ACh) to move toward and fuse with the presynaptic membrane, releasing their contents into the synaptic cleft by exocytosis.
Acetylcholine then diffuses rapidly across the narrow cleft and binds to nicotinic receptors on the muscle end plate. These receptors are ligand-gated ion channels, meaning they open when ACh binds. Once opened, sodium ions flow into the muscle fibre, producing a local depolarisation called the end-plate potential. This depolarisation is not yet a muscle action potential, but it brings the muscle membrane closer to the threshold required to fire one.
If the end-plate potential is large enough, it triggers a muscle action potential that spreads across the sarcolemma and down the T-tubules. This electrical signal then stimulates the sarcoplasmic reticulum to release calcium into the cytoplasm, linking the nerve impulse to the contractile machinery of the muscle. This conversion of electrical activity into mechanical force is what allows a neural signal to produce visible movement.
Termination of the Signal and Safety of Transmission
Neuromuscular transmission must be brief and precisely controlled so muscles can contract and relax rapidly. Acetylcholine is quickly broken down in the synaptic cleft by the enzyme acetylcholinesterase, producing acetate and choline. Choline is actively transported back into the motor neuron terminal, where it is recycled to make new ACh. This rapid removal prevents continuous stimulation of the muscle fibre, allowing it to relax and be ready for the next impulse.
The neuromuscular junction also operates with a built-in safety factor. Each nerve impulse releases more ACh than is strictly required to trigger a muscle action potential. This means that even if some receptors are blocked or some ACh is lost, transmission remains reliable. This redundancy is why healthy neuromuscular transmission is so dependable, and why weakness only appears when a significant number of receptors are damaged or ACh release is impaired, as occurs in disorders such as myasthenia gravis.
Integration With Excitation–Contraction Coupling
The neuromuscular junction is the gateway between the nervous system and the muscle fibre, but it does not directly cause contraction. Instead, it initiates the electrical events that activate the muscle’s internal machinery. Once the muscle action potential travels through the T-tubules, it triggers calcium release from the sarcoplasmic reticulum.
The released calcium binds to troponin, causing tropomyosin to move away from the myosin-binding sites on actin. This allows cross-bridge cycling to begin, generating force and shortening the muscle fibre. In this way, the NMJ serves as the crucial first step that links nerve activity to muscle contraction, ensuring that voluntary movement is fast, precise, and repeatable.
Clinical Connections
Disorders of the neuromuscular junction produce characteristic patterns of weakness and paralysis because they interfere with the highly reliable process of signal transmission between nerve and muscle. In myasthenia gravis, autoantibodies destroy or block acetylcholine receptors on the muscle end plate, reducing the safety factor of transmission. Each nerve impulse releases normal amounts of acetylcholine, but there are too few functioning receptors to reliably generate a muscle action potential. With repeated use, acetylcholine stores are temporarily depleted and muscle contractions become progressively weaker, producing the classic fatigable weakness that improves after rest.
In botulism, the bacterial toxin prevents acetylcholine release from presynaptic nerve terminals, so the muscle fibre never receives the signal to contract. This causes flaccid paralysis that can progress to respiratory failure if the diaphragm and intercostal muscles are affected. In contrast, organophosphate poisoning blocks acetylcholinesterase, allowing acetylcholine to accumulate and overstimulate the end plate. This produces continuous depolarisation with muscle twitching, cramps, and eventually paralysis when the muscle can no longer respond to sustained stimulation.
Disruption of neuromuscular junction transmission produces distinct clinical effects depending on how acetylcholine signalling is altered, including:
Myasthenia gravis: reduced ACh receptors, fatigable weakness
Botulism: blocked ACh release, flaccid paralysis
Organophosphate poisoning: excessive ACh, fasciculations and paralysis
Neuromuscular blockers: intentional interruption of NMJ transmission
In anaesthesia and critical care, neuromuscular blocking agents are deliberately used to interrupt transmission at the NMJ to produce controlled muscle relaxation for surgery, intubation, and mechanical ventilation. These drugs either block acetylcholine receptors or prevent depolarisation, temporarily paralysing skeletal muscles while leaving consciousness and pain perception unchanged. This is why patients receiving neuromuscular blockers require full sedation and ventilatory support.
Recognising early signs of neuromuscular failure, such as ptosis, dysphagia, or declining respiratory effort, is essential to safely monitor patients receiving paralytic drugs or at risk of toxin exposure. Accurate assessment and timely intervention are often lifesaving in conditions that disrupt neuromuscular transmission.
Concept Check
Why is calcium entry into the presynaptic terminal essential for neurotransmitter release?
What makes the neuromuscular junction more reliable than most other synapses?
Why does blocking acetylcholinesterase initially increase muscle contraction but later cause paralysis?
Why does myasthenia gravis cause weakness that worsens with activity?
Where does the NMJ fit within excitation–contraction coupling?